The practice of medicine has been transformed by the introduction by a series of antifungal azole agents starting with ketoconazole in 1985, but spurred on by fluconazole, itraconazole and voriconazole over the following 2 decades. Information about availability and pricing in different countries can be viewed on the GAFFI website.
Patient outcomes were transformed and oral therapy became possible. The low toxicity of these agents compared with amphotericin B was transformational. Many different topical azoles are available clinically for thrush (yeast infection) but not all are included here.
Azole antifungals interfere with fungal biosynthesis of ergosterol, by inhibiting 14-α demethylase. For more information about this pathway and how resistance can develop, please see Doctor Fungus or reviews by Cowen et al (2015) or Shafiei et al (2020).
Therapeutic drug monitoring (TDM) is recommended when prescribing voriconazole or itraconazole to certain patient groups. Find out more by watching our TDM webinar and Q&A on YouTube.
Azoles interact with many other drugs and may require dose adjustments to be made. Find out more using the Antifungal Drug Interactions database and app produced by the Fungal Infection Trust.
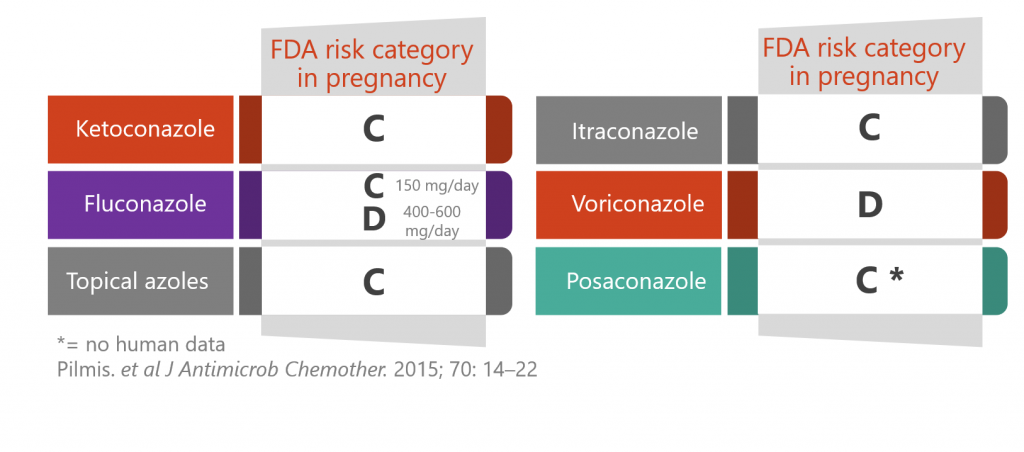
Prescribing azoles during pregnancy
Oral azoles have harmful effects on a foetus by interfering with steroidogenesis. However, in some cases human trial data are lacking (category C) and in other cases their use may still be indicated for a severe/invasive fungal infections (category D). Lipid formulations of amphotericin B are generally a safer option during pregnancy.
Find out more by watching our clinical lecture on using antifungals during pregnancy and breastfeeding.
- Fluconazole can be considered after the first trimester, in the absence of a suitable alternative
- Voriconazole is contra-indicated during pregnancy
- Effective contraception is vital during (and for 2 months after) treatment with itraconazole
Clinical lectures
Click here to download the slide decks for the lectures
Azole resistance
Azole resistance is becoming more common (perhaps due to heavy use of azoles in agriculture) so it is important to carry out susceptibility testing when possible, either by the CLSI microdilution method or using molecular methods to detect mutations in genes such as cyp51A.
Resistance may also arise in patients on long-term therapy for infections such as CPA. Antifungal stewardship programmes can help to guide usage to minimise this.
- Direct detection of some resistance mutations in Aspergillus fumigatus is possible with PCR (White et al, 2017; Dannaoui et al, 2017)
- Read about how researchers at MRCM used pyrosequencing to detect azole resistance in Aspergillus fumigatus strains isolated from CPA/ABPA patients at the National Aspergillosis Centre (van der Torre et al, 2020)
Factsheets
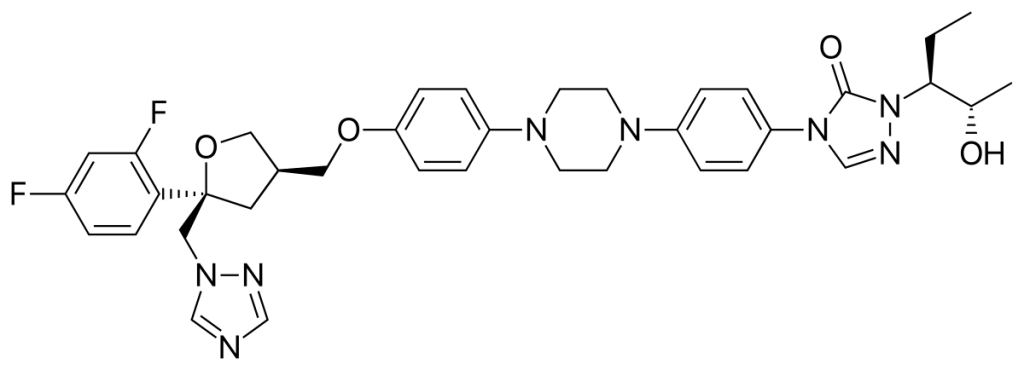
Posaconazole
| NAME(S) Posaconazole Noxafil (Merck) |
| APPROVAL 2006, for treatment of 2nd-line treatment of invasive aspergillosis/mucormycosis, and for prophylaxis in acute leukaemia and following HSCT. Look up the label and prescribing information |
| STRUCTURE & MECHANISM Synthetic fat-soluble triazole. It interferes with fungal biosynthesis of ergosterol like all azoles, by inhibiting 14-α demethylase. |
| FORMULATIONS & DOSES Posaconazole is formulated as an oral solution containing numerous ingredients including polysorbate 80 and cherry flavouring, also as tablets and as an intravenous product. The oral solution is better absorbed if it is taken with food because this increases the absorption through the gut wall, but the tablet can be taken at any time. The standard dose of oral solution of posaconazole dose is 300 mg twice daily, then 300 mg once daily thereafter. In patients taking posaconazole prophylaxis oral tablets – the normal dose is 300 mg twice a day for first day, then 300 mg once daily. The concentration of the oral suspension is 40 mg/ml, so a 5 ml spoon delivers 200 mg. For posaconazole tablets and intravenous formulation the standard daily dose is 300 mg once daily (after the initial day at 300 mg 2x day). |
| ACTIVITY Extremely broad spectrum. – ACTIVE against Aspergillus spp., Candida spp., Coccidioides spp., Histoplasma, Paracoccidioides brasiliensis, Blastomyces dermatitidis, Cryptococcus spp., Sporothrix schenckii, various species of Mucorales and Fusarium spp. and numerous other black moulds such as Bipolaris spp. and Exserohilum spp. The majority of Aspergillus isolates are killed in test tube tests by posaconazole at clinically relevant concentrations; a higher proportion than any other antifungal agent. – ACQUIRED RESISTANCE to posaconazole does occur in Aspergillus fumigatus and Candida albicans but is otherwise rare. |
| TYPICAL REGIMEN Treatment of refractory fungal infections, aspergillosis or zygomycosis, or other rare infection – 300 mg twice daily, then 300 mg once a day thereafter. Monitoring of blood levels of posaconazole shows ~10-fold variation in levels, and low levels are associated with failure. (Dose escalation to 400mg three times daily might be helpful, if the reason for low levels is not a drug interaction). For prophylaxis of fungal infection, 300mg IV, twice daily for first day, followed by 300mg IV, once day thereafter. Oral tablets 300 mg twice daily for first day and thereafter 300 mg once daily. For oral suspension 200 mg 3x per day. For oropharyngeal candidiasis, 100 mg oral suspension twice daily, followed by 100 mg once daily. For treatment when refractory to itraconazole or fluconazole 400 mg twice daily oral suspension. |
| PHARMACOKINETICS Posaconazole is absorbed over three to five hours. Increasing doses from 200-800 mg daily yields a proportional increase in exposure. It takes 7-10 days to reach steady state levels in blood but most people have sufficient concentrations for activity within two days. Posaconazole has a very large apparent volume of distribution suggesting extensive tissue uptake. It is also very highly protein bound (>98%). Posaconazole is metabolised primarily by the liver into glucuronide conjugates. The elimination half life is typically 35 hours with most of the drug being excreted in the faeces as unchanged drug. There is considerable patient to patient variation in drug absorption and exposure, and some transplant patients have consistently low plasma levels. |
| DRUG INTERACTIONS Posaconazole has fewer drug interactions than itraconazole or voriconazole. Plasma levels fall when posaconazole is co-administered with enzyme reducers such as rifampicin, rifabutin, efavirenz, phenytoin, phenobarbitone, carbamazepine and others. Posaconacole is less well absorbed when it is given in association with PPIs (omeprazole, lansoprazole) or H2 receptor antagonists (cimetidine, ranitidine etc). View drug interaction database. Posaconazole is a potent inhibitor of CYP3A4. This means that the blood levels of compounds that are metabolised through this system are likely to rise. Important examples include cyclosporine, tacrolimus and sirolimus, terfenadine, astemizole, and cisapride, ergotamine for migraine, certain statins including simvastatin, lovastatin and atorvastatin, vincristine or vinblastine chemotherapy, midazolam, diazepam and other benzodiazapines and some HIV protease inhibitors. Other drugs with minor interaction with posaconazole include digoxin and glipizide for diabetes and certain calcium channel blockers such as nifedipine and diltiazem. Find out more using the Antifungal Drug Interactions database and app produced by the Fungal Infection Trust. |
| SIDE EFFECTS & TOXICITY Common side effects include nausea, mild diarrhoea and abdominal discomfort, indigestion and dry mouth. Liver function tests may also become elevated with the same frequency as with fluconazole. Skin rashes due to posaconazole are relatively common. Fever, fatigue and lacking in energy and loss of appetite is also moderately common. Numerous other side effects such as low white blood cells or platelets were apparently associated with posaconazole because the drugs were studied in neutropenia and leukaemia therapy. The frequency of such events in patients only treated with posaconazole without underlying leukaemia is low. |
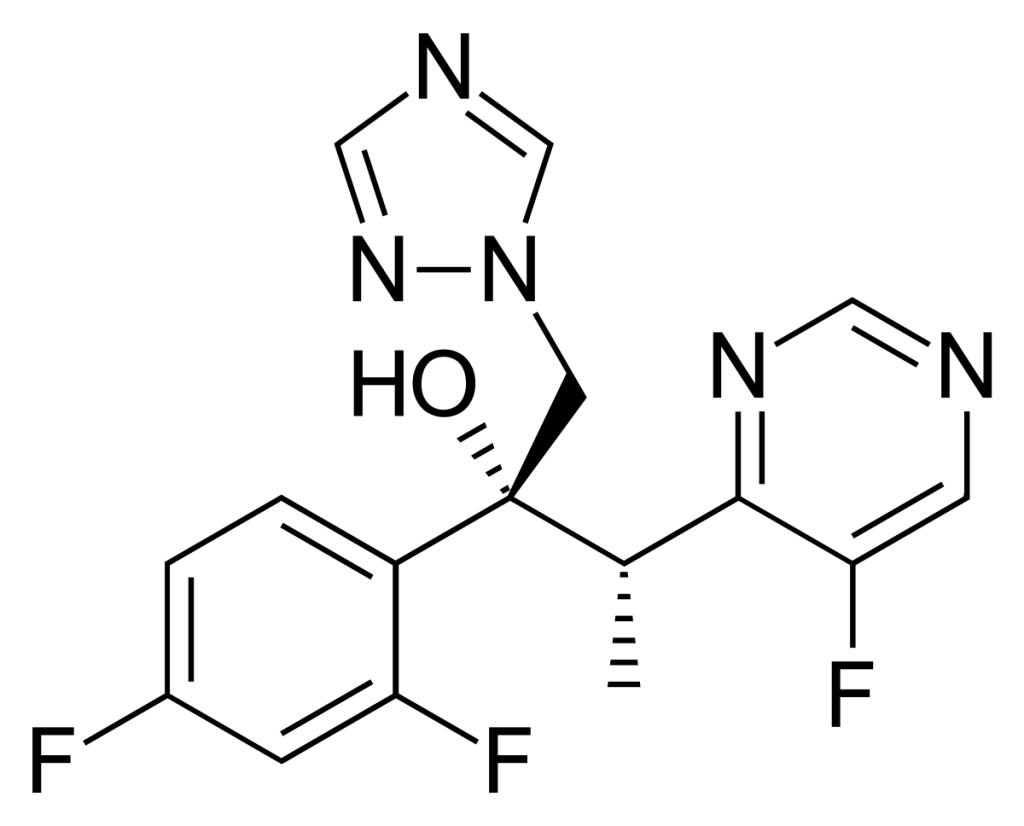
Voriconazole
| Names | NAMES Voriconazole V-fend (Pfizer); voriconazole Accord (Accord Healthcare). |
| Approval | APPROVAL 2002, for treatment of invasive aspergillosis. Look up the label and prescribing information Availability and pricing |
| Structure & mechanism | STRUCTURE & MECHANISM 2nd-generation triazole. It interferes with fungal biosynthesis of ergosterol like all azoles, by inhibiting 14-α demethylase. |
| Dose & delivery | FORMULATION Oral, intravenous and ocular preparations of voriconazole are available. For intravenous usage a loading dose of 6 mg/kg twice at 12 hour intervals is recommended for life threatening infections in adults, followed by 4 mg/kg twice daily. The dose in children should be 7 mg/kg twice daily intravenously, although very young children may require proportionately larger doses because of rapid clearance. If drug-monitoring facilities are available the dose can be altered on the basis of blood levels. Bioavailability is nearly 100% so oral dosing can be relied upon, which is usually 200 mg twice daily. Lower treatment doses should be given to adults weighing less than 40 kg, elderly people and those with prior liver disease to minimise the chance of high levels and toxicity. Dose increases result in disproportionate increase in drug exposure in adults, but not children, because the pharmacokinetics of voriconazole is not linear in adults. |
| Active against | ACTIVITY Broad spectrum. ACTIVE against the vast majority of Candida spp., Cryptococcus spp., all Aspergillus spp., Scedosporium apiospermum, some isolates of Fusarium, and many rather rare pathogens. NOT ACTIVE against Mucorales species such as Mucor spp., Rhizopus spp., Rhizomucor spp., and others. |
| Typical regimen | TYPICAL REGIMEN For invasive aspergillosis: 6 mg/kg twice (loading), followed by 3-4 mg/kg twice daily, followed by 200 mg twice daily orally, in adults; proportionally larger doses in children. |
| Metabolism & excretion | PHARMACOKINETICS Voriconazole is metabolised by the liver. The most important enzymes involved in this metabolism are CYP2C19, CYP3A4 and CYP2C9. Genetic variation between different individuals, particularly with CYP2C19, partially affects the rate at which the drug is metabolised. Slow metabolisers are found in about 3% of Caucasians and in approximately 20% of patients from Japan and other parts of Asia; these patients may accumulate drug and sometimes very high concentrations are found in the blood. Elderly people and those with moderate or severe liver disease are also slow metabolisers. It is necessary to increase the dose in children because young children appear to metabolise the drug very fast, and older children and teenagers quite fast. |
| Drug interactions | DRUG INTERACTIONS As with most azoles, many drugs interact with voriconazole. – REDUCE THE ACTION OF VORICONAZOLE: rifampicin, refabutin (less marked), phenytoin and carbamazapine. – REDUCE METABOLISM OF VORICONAZOLE: omeprazole and probably other proton pump inhibitors. – VORICONAZOLE INCREASES THE ACTION OF : some antihistamines (terfenadine and astemizole), prednisolone, sirolimus, cyclosporin (halve the dose), tacrolimus (reduce dose to one third), ergotamine, warfarin, sulphonylureas, statins, midazolam and other benzodiazepines, vincristine and some HIV drugs. Find out more using the Antifungal Drug Interactions database and app produced by the Fungal Infection Trust. |
| Side effects | SIDE EFFECTS & TOXICITY Perhaps the most common side effect associated with voriconazole is visual side effects. Patients describe wavy lines, mild blurred vision or bright lights in the first few days after taking voriconazole, which typically lasts a few minutes. Extensive investigations have not unearthed the precise mechanism but the side effect wears off over time and only rarely has their been any long term damage to the eye. Some patients develop abnormal liver function tests with voriconazole and if these are remarkably elevated then the drug should be stopped. High dosages result in reduced concentration and a sensation of a ‘muzzy head’, and sometimes hallucinations and frank confusion. Peripheral neuropathy also occurs after some months of therapy in a small minority of patients. Occasional serious side effects have occurred in patients with very high concentrations of voriconazole. The long term usage of voriconazole may lead to reddening of exposed skin, especially the face, dry eyes and dryness of the lips, in those with white skin. White patients should wear sunscreen (SPF50) and avoid sunny environments such as conservatories. UV protective film can be used on car windows. |
Itraconazole
| NAMES Itraconazole Sporanox (Janssen); TOLSURA/Lozanoc (Mayne Pharma) |
| APPROVAL 1991 It is now off patent, although the IV and oral solution formulations are still patent protected Look up the label and prescribing information Availability and pricing |
| STRUCTURE & MECHANISM It is a fat soluble synthetic triazole antifungal drug. It interferes with fungal biosynthesis of ergosterol, by inhibiting 14-α demethylase. |
| FORMULATIONS & DOSAGES Until 1997, only capsules were available, which are better absorbed taken with food. There is some variation in bioavailability between different capsule formulations, and switching between them has lead to clinical failure. An oral liquid form was then introduced, particularly for the management of fluconazole-resistant oral thrush in patients with AIDS. Also used for prophylaxis in leukaemia or after HSCT, and for patients who are on acid reducing drugs. Suspension is better absorbed taken on an empty stomach. An IV formulation in cyclodextrin is also licensed, and is used when patients are unable to take oral medication or are very ill. SUBA-itraconazole is a recent formulation that uses a polymer matrix to increase bioavailability (read more at Mayne Pharma) |
| ACTIVITY Particularly broad spectrum. – ACTIVE against Aspergillus spp., Blastomyces dermatitidis, Candida (all species and most isolates including some resistant to fluconazole), Coccidioides spp., Cryptococcus spp., Histoplasma spp, Paracoccidioides brasiliensis, Scedosporium apiospermum and Sporothrix schenckii. It is also active against all skin fungi (dermatophytes). – NOT ACTIVE against Mucorales or Fusarium spp. and a few other rare fungi. Until the development of posaconazole, it was the most active against black moulds, including Bipolaris, Exserohilum. Resistance to itraconazole is described in Candida spp. and in Aspergillus fumigatus and A. niger. |
| TYPICAL REGIMENS Candida: Two doses of itraconazole (200 mg) are sufficient for most cases of vaginal thrush. Five to seven days of therapy are sufficient for oral thrush. Occasionally in AIDS continuous therapy is required for fluconazole resistant disease. Skin: Seven days of 200 mg itraconazole daily is usually used for most skin infections, except for extensive tinea pedis (fungal infection of the feet and between the toes) and some scalp infections in which case 100 mg daily for 3-4 weeks therapy is used. Three months of therapy (200 mg daily) are used for fingernail disease and 6 months for toenail disease, sometimes in interrupted regimens (so called pulse therapy ( 200 mg twice daily for 7 days, repeated 3 weeks later). INVASIVE INFECTIONS: Loading doses of 200 mg 3 or 4 times a day for 3 to 4 days are appropriate. For invasive aspergillosis, typically 400 mg per day or more are used for periods of at least 3 months, and often as long as a year or more. ABPA: 400 mg appears to be slightly superior to 200 mg. A minimum of 3 months therapy is required and relapse is common after discontinuation – therefore a longer, even continuous, course of therapy may be appropriate. CPA: itraconazole is not very effective, but 400 mg per day for many months is partially effective. TDM is appropriate to ensure adequate levels and compliance, and minimise toxicity. Histoplasmosis, paraccoidioidomycosis, blastomycosis and sporotrichosis: 200 mg per day given for 3-6 months is effective in the vast majority of cases. For AIDS patients larger doses are used for these infections, typically 400 mg per day and treatment may be lifelong, although a dose reduction to 200 mg per day after control of disease is appropriate. In coccidioidomycosis 400 mg per day appears to be the best dose. SINUS: For black mould infections, substantially larger doses of 400-1200 mg per day have been used, but specialist advice is necessary. |
| PHARMACOKINETICS Absorption from the gut is incomplete. It is extensively metabolised, and one metabolite (hydroxyitraconazole) has antifungal activity. The drug is extensively protein bound and therefore does not pass into urine or CSF. However, it does bind avidly to keratin and so high concentrations are found in the skin and nails. The half life itraconazole in the blood varies with dosage but is typically 48 to 60 hours, and it takes 2 weeks for blood levels to reach a steady state. If loading doses are used, steady state can be achieved in a week. |
| DRUG INTERACTIONS Numerous drugs interact with itraconazole, which is one of its main limitations. – REDUCE ACTION OF ITRACONAZOLE: The absorption of capsules is reduced if given with antacid drugs, but increased if the solution formulation is used. If enzyme inducing drugs such as rifampicin (rifampin), phenytoin or carbamazepine are used, there is a marked reduction in the blood levels of itraconazole and treatment may fail. – ITRACONAZOLE INCREASES ACTION OF: drugs including terfenadine, astemizole and cisapride, as it prevents them from being metabolised, which can lead to serious heart arrhythmias. These drugs should not be given with itraconazole. Certain sedatives will have prolonged action if given with itraconazole, in particular midazolam and triazolam. Itraconazole may also increase the blood levels of digoxin, cyclosporin and warfarin and these drugs and their effects should be carefully monitored. Anticancer therapy with the drug vincristine given together with itraconazole, may have prolonged action and cause neurological damage, which is usually temporary but can be very problematic. Interactions with inhaled steroids can be profound, leading to permanent adrenal failure. Find out more using the Antifungal Drug Interactions database and app produced by the Fungal Infection Trust. |
| SIDE EFFECTS & TOXICITY Itraconazole is usually tolerated adequately. The commonest side effects are nausea and sickness, occasional abdominal discomfort and constipation. These, and diarrhoea, are more common with itraconazole solution. In older people fluid retention can be a problem. Rashes occur in approximately 1 in 20 patients and occasionally are severe. Abnormal liver function tests occasionally occur and in rare cases severe liver disease or jaundice. Occasionally adrenal gland suppression, low blood potassium and raised blood pressure can occur. Peripheral neuropathy and tremor are also problematic in some patients |

Fluconazole
| NAMES Fluconazole Diflucan (Pfizer) |
| APPROVAL 1990, went off patent in 2003. Look up the label and prescribing information Availability and pricing |
| STRUCTURE & MECHANISM A bis-triazole It interferes with fungal biosynthesis of ergosterol like all azoles, by inhibiting 14-α demethylase. |
| FORMULATIONS & DOSAGES The drug is available as a capsule, oral suspension and intravenous infusion. Candida: A typical dose for oral thrush is 100 mg per day. The typical dose for vaginal thrush is a 150 mg per day. For the prevention of fungal infections in leukaemia, doses from 50-400 mg per day have been used and studied; most authorities recommend between 100-400 mg. For candidaemia and other serious Candida infections a minimum dose of 400 mg per day (after a loading dose of 800 mg) is recommended, with increased doses in patients on haemofiltration and/or rifampicin (see below). Some authors have suggested that 800 mg is superior for candidaemia, but the data is not compelling. Cryptococcal meningitis: after amphotericin B, a minimum of 400 mg per day initially is used until the patient is stable and then typically 200 mg per day. Much larger doses e.g. 800-1200 mg per day should be used as an alternative primary regimen (preferably with flucytosine) or for patients failing therapy. Coccidioidal meningitis: larger doses such as 800-1200 mg per day have been used. Almost all other treatments have been for 100-400 mg per day. Reduced doses may be appropriate in severe renal dysfunction. |
| ACTIVITY – ACTIVE against most Candida species, with the absolute exception of Candida krusei and partial exception of Candida glabrata and a small number of isolates of Candida albicans, Candida tropicalis, Candida parapsilosis and other rare species. It is also active against the vast majority of Cryptococcus spp. isolates. It is active against many other yeasts including Trichosporon spp., Rhodotorula rubrum, and the dimorphic endemic fungi including Blastomyces dermatitidis, Coccidioides spp., and Histoplasma spp. It is less active than itraconazole against these dimorphic fungi. – NOT ACTIVE against Aspergillus spp. or Mucorales. It is active against skin fungi such as Trichophyton spp. – RESISTANCE: some documented in Candida albicans, especially oral isolates in patients with AIDS previously treated with fluconazole. Typical rates of resistance, in Candida albicans in a general hospital are 3-6%. |
| PHARMACOKINETICS It is water soluble and well absorbed after oral administration, and serum concentration parallels dose. The drug penetrates well into cavities such as around the brain, the eyes, saliva and urine. Most of the drug is excreted in the urine; very little is metabolised by the liver. Babies and children, especially those with fevers, have accelerated metabolism. A maximum 17% of the dose is excreted in breast milk in lactating women, with minimal or no side effects in babies exposed to it. |
| DRUG INTERACTIONS Few drug interactions. REDUCE THE ACTION OF FLUCONAZOLE: the most profound is with rifampin (rifampicin) and phenytoin, which accelerate the metabolism of fluconazole in liver and reduce serum levels. FLUCONAZOLE INCREASES THE ACTION OF: phenytoin and diabetic drugs such as chlorpropamide, glibenclamide, glipizid, tolbutamide and warfarin, via a rise in serum concentration. It also increases cyclosporin levels slightly. Find out more using the Antifungal Drug Interactions database and app produced by the Fungal Infection Trust. |
| SIDE EFFECTS & TOXICITY Generally well tolerated, and probably the least toxic of all the systemic antifungal drugs. Common side effects are nausea and abdominal discomfort; raised liver function tests occasionally occur. Skin rashes occur in up to 1 in 20 patients and these may be severe in rare cases. There are no endocrine effects. Fluconazole should not be given in pregnancy. |
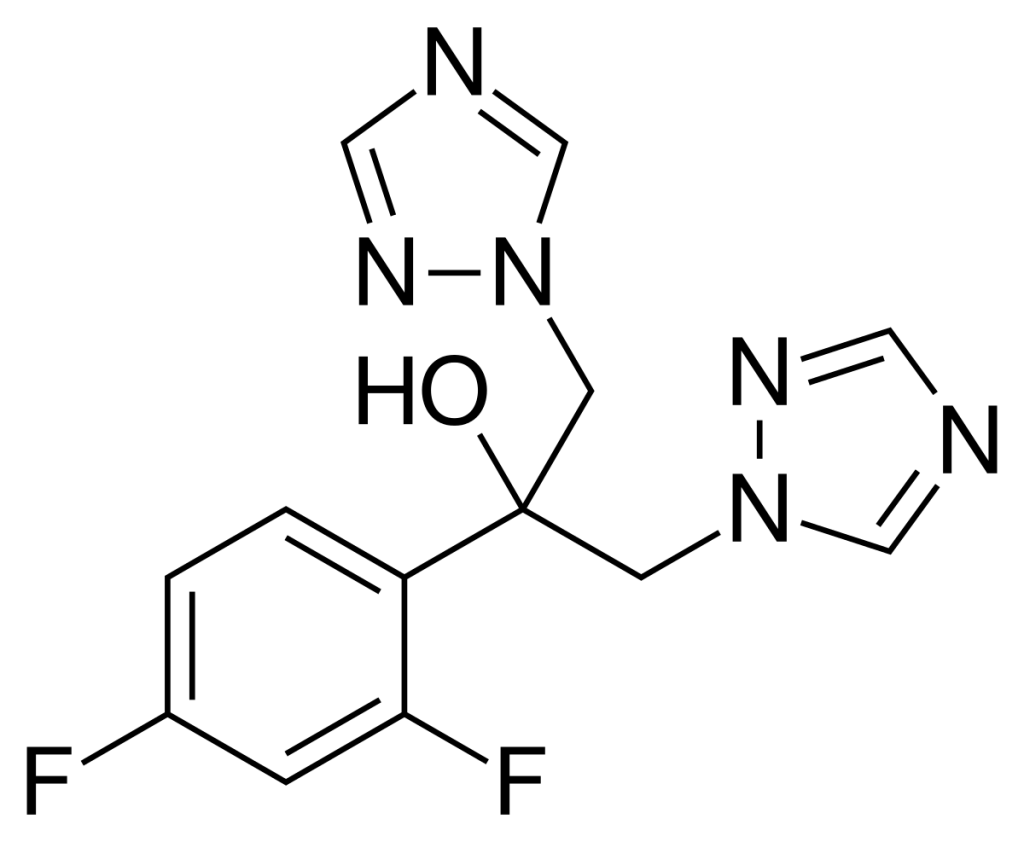
Ketoconazole
| NAMES Ketoconazole |
| APPROVAL 1985 The first orally active azole compound. N.B. In 2013 the FDA restricted the use of ketoconazole to life-threatening infections and endemic mycoses not responding to other drugs as other antifungals are superior and carry less risk of toxicity (particularly to the liver). Look up the label and prescribing information |
| STRUCTURE & MECHANISM A synthetic imidazole. It interferes with fungal biosynthesis of ergosterol like all azoles, by inhibiting 14-α demethylase. |
| FORMULATIONS & DOSAGES Available as oral tablets or as a topical preparation such as Nizoral shampoo. |
| ACTIVE against Candida spp, Blastomyces dermatitidis, Coccidioides spp, Histoplasma and Paracoccidioides. It is also active against skin fungi, but not to the degree of itraconazole or terbinafine. NOT ACTIVE against Aspergillus spp. |
| TYPICAL REGIMEN Candida: used for the treatment of oral thrush in a dose of 200 mg per day, or 400 mg per day in patients with AIDS and resistant strains. Blastomycosis, histoplasmosis, coccidioidomycosis, paracoccidioidomycosis: doses of 400 mg per day have been used (given as 200 mg twice a day, but it is inferior to itraconazole for these indications). The same dose is used for skin infections. As ketoconazole is not as active as itraconazole, longer courses of therapy are typically given for endemic mycoses. |
| PHARMACOKINETICS Ketoconazole is well absorbed after oral administration with or without food. Almost all the drug is metabolised in the liver and the metabolites are excreted in bile. |
| DRUG INTERACTIONS – REDUCE THE ACTION OF KETOCONAZOLE: levels are slightly lower in those on anti-ulcer drugs. Rifampicin (rifampin) reduces the concentration of ketoconazole in the blood because of accelerated metabolism. – KETOCONAZOLE INCREASES THE ACTION OF: terfenadine, astemizole and cisapride and must not be combined with them, as this can cause abnormal heart rhythms (prolonged Qt interval). Ketoconazole also can prolong the action of certain sedatives such midazolam and triazolam. It increases the blood level of cyclosporin and in fact has been used in some transplant centres to reduce the cost of cyclosporin. Ketoconazole also prolongs the effect of warfarin. Find out more using the Antifungal Drug Interactions database and app produced by the Fungal Infection Trust. |
| SIDE EFFECTS & TOXICITY Loss of appetite, feeling sick and vomiting are the most common side effects and occur in about 1 in 10 patients. The side effects are particularly a problem with high doses (>400 mg per day). Ketoconazole had caused severe liver reactions, about 1 in 20 patients get slight elevations of liver function tests on ketoconazole. If the treatment is given for longer than two weeks there is a rare side effect of severe liver disease, carrying a 1-10,000 – 15,000 risk, which is fatal if not diagnosed and ketoconazole stopped. Ketoconazole also inhibits human adrenal and testicular steroid synthesis, particularly at high doses. This has consequences of loss of hair, breast swelling and lack of sexual function. There is a possibility that these effects may be more problematic in teenagers during puberty and ketoconazole should be avoided in this age group if possible. Ketoconazole should not be given in pregnancy. Ketoconazole is used for controlling corticosteroid excess in Cushing’s disease and is quite effective, typically at 600 mg daily. |
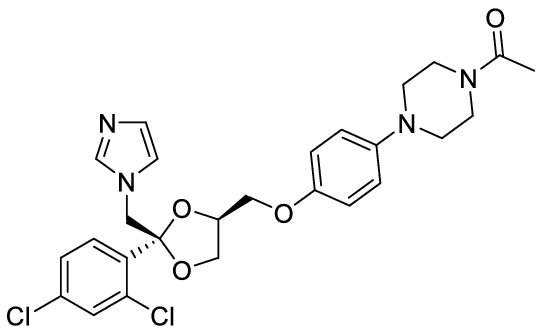
Econazole
| NAMES Econazole Ecostatin, Ecoderm, Pevaryl and multiple other names |
| APPROVAL 1974 Look up the label and prescribing information |
| STRUCTURE & MECHANISM A synthetic imidazole. It interferes with fungal biosynthesis of ergosterol, by inhibiting 14-α demethylase, but may also have other cellular actions based on studies in Candida albicans. |
| FORMULATIONS & DOSAGES Econazole 1% cream, lotion, powder or solution is only used topically usually 3 times daily for 2-4 weeks. Econazole is effective for seborrhoeic dermatitis, nappy rash due to Candida, some cases of ringworm, tinea cruris and athlete’s foot. A 1% solution can be used for otitis externa. For inflammatory conditions such as seborrhoeic dermatitis, combined preparations with topical corticosteroid are helpful. Vaginal candidiasis: pessaries of 150 mg are give daily for 3 nights. Alternatively 5 g of a 1% vaginal cream can be given for 2 weeks each night. Econazole cream is also used to treat penile candidiasis (balanitis). |
| ACTIVITY – Active against most Candida species and skin fungi such as Trichophyton spp., Microsporum spp., Epidermophyton floccosum and Malassezia spp., as well as some systemic fungal pathogens such as Aspergillus spp. It has some anti-bacterial activity as well. – RESISTANCE: some is documented in Candida albicans, and can be inferred from miconazole susceptibility results. |
| PHARMACOKINETICS Systemic absorption of econazole is minimal when applied to the skin as it penetrates only the surface layer (epidermis). Vaginal administration leads to minimal systemic absorption, but may be enough to initiate an attack of acute intermittent porphyria in those with this rare disease. |
| DRUG INTERACTIONS Very few drug/drug interactions with econazole. Pessaries of econazole may damage latex condoms or diaphragms and so alternative contraceptive precautions are best used |
| SIDE EFFECTS & TOXICITY Econazole may lead to local skin irritation and a burning sensation. Econazole may be given in pregnancy, although nystatin is preferred. |
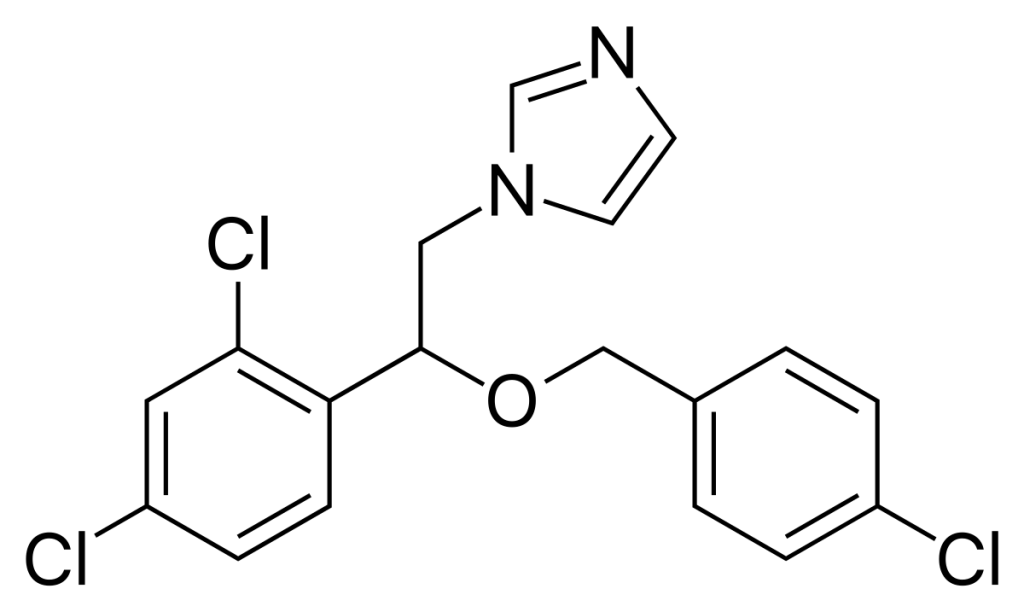
Clotrimazole
| NAMES Clotrimazole Canesten (Bayer) |
| STRUCTURE & MECHANISM Synthetic imidazole |
| APPROVAL 1982 Became over-the-counter in USA in 1989 Look up the label and prescribing information |
| FORMULATIONS & DOSAGES Clotrimazole is only used topically, and is formulated alone or with other active ingredients such as corticosteroids. Clotrimazole is effective for seborrhoeic dermatitis, nappy rash due to Candida, some cases of ringworm, tinea cruris and athlete’s foot. A 1% solution can be used for otitis externa. For inflammatory conditions such as seborrhoeic dermatitis, combined preparations with topical corticosteroid are helpful. |
| ACTIVITY – ACTIVE: against most Candida species and skin fungi such as Trichophyton spp., Microsporum spp. and Epidermophyton floccosum, Malassezia spp. In vitro it also has activity against many fungi generally susceptible to azoles such as Coccidioides immitis, but as it cannot be given systemically, this is of limited importance. – RESISTANCE: some is documented in Candida albicans, especially among oral isolates in patients with AIDS treated with Clotrimazole. Typical rates of resistance, in Candida albicans in a general hospital are 3-6%. |
| TYPICAL REGIMEN Most topical skin preparations are 1% cream, lotion or spray, which should be applied 2 or 3 times daily for 2-4 weeks. For vaginal candidiasis, pessaries of 100 mg are give daily for 6 days, 200 mg for 3 days and 500 mg as a single dose. Alternatively 1%, 2% or 10% vaginal cream delivering roughly the same doses as pessaries, may be used, for the same duration. Clotrimazole cream is also used to treat penile candidiasis (balanitis). Clotrimazole 10 mg lozenges or troches are used to treat oral candidiasis. They are taken 5 times daily for 14 days for treatment, or 3 times daily for prevention of oral candidiasis. |
| PHARMACOKINETICS Clotrimazole is practically insoluble in water. Applied on the skin it penetrates on the surface layer (epidermis), with no systemic absorption. Between 3-10% is absorbed systemically after vaginal administration. Clotrimazole is metabolised by the liver. |
| DRUG INTERACTIONS There are very few drug interactions with clotrimazole. Pessaries may damage latex condoms or diaphragms, so additional contraceptive measures are wise. |
| SIDE EFFECTS & TOXICITY Generally well tolerated but nausea, vomiting, unpleasant mouth and pruritus (itching) have been reported with oral lozenges. Local skin irritation and a burning sensation are reported after use on the skin. Clotrimazole may be given in pregnancy, although nystatin is preferred. |
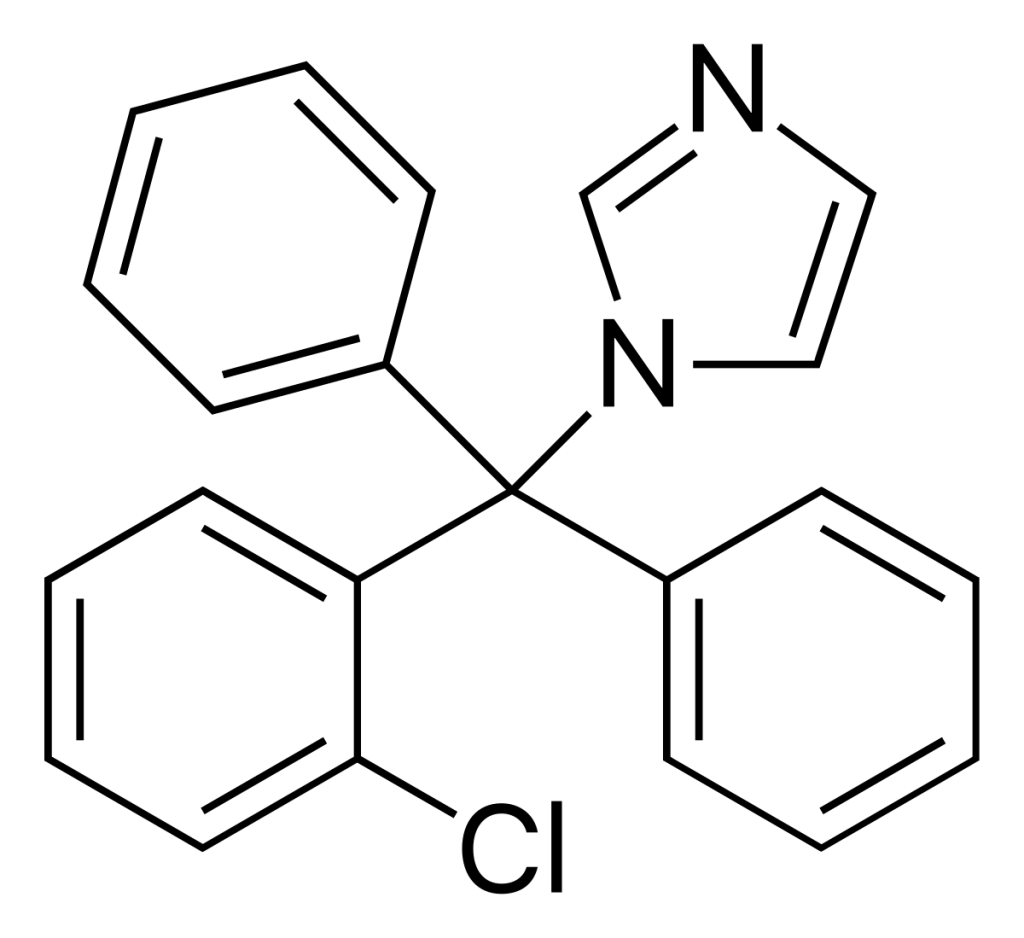
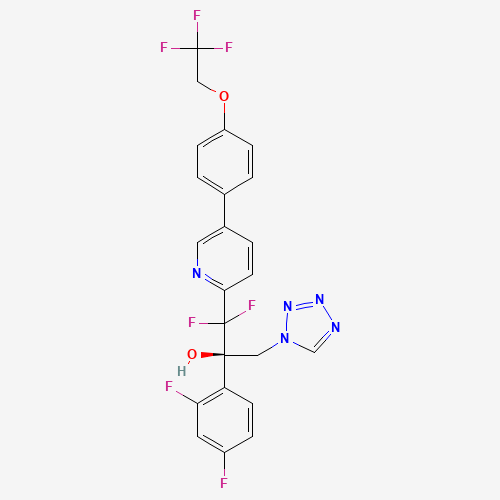
Oteseconazole
| NAME(S) Oteseconazole (Vivjoa) |
| APPROVAL Discovered in 2013 as VT-1161 by Viamet Pharmaceuticals. Approved for use in the United States in 2022 for treatment of recurrent vulvovaginal candidiasis, by Mycovia Pharmaceuticals. |
| STRUCTURE & MECHANISM Oteseconazole is the first of a new class of tetrazole antifungals. Their mode of action is almost the same as triazoles (inhibiting ergosterol synthesis) but they have more selectivity for the fungal enzyme. |
| FORMULATIONS & DOSES Oral capsules of 150mg. |
| ACTIVITY ACTIVE – most Candida spp., including azole resistant C. albicans and C. krusei and echinocandin resistant C. glabrata and C. auris. Rhizopus spp. Trichopyton spp. Microsporum spp. Coccidioides spp. Cryptococcus spp. Histoplasma spp. Blastomyces spp. Reduced susceptibility or resistance may be mediated by upregulation of the efflux pumps CDR1, MDR1, and the azole target, lanosterol 14-alpha-demethylase (CYP51). INACTIVE – Aspergillus spp. |
| TYPICAL REGIMEN Prevention of recurrence of recurrent vulvovaginal candidiasis: Dosing requires two loading doses of 600mg on day 1 and 450 mg on day 2, then 150mg once a week, for up to 11 weeks. An alternative regimen using oteseconazole and fluconazole in combination is also used. |
| PHARMACOKINETICS Oteseconazole is slowly absorbed over 5-10 hours, and absorption increased with a fatty meal. It has a very high volume of distribution, protein binding of >99%, a long half-life of 138 days and is not metabolised at all. Peak concentrations occur after 6-9 hours and the terminal half-life is 138 days. It is extensively distributed to most tissues in much greater concentrations than in plasma (4- to 50-fold), but not to the brain. Oteseconazole does not undergo significant metabolism. |
| DRUG INTERACTIONS Oteseconazole has few documented drug interactions. It is a BCRP (Breast Cancer Resistance Protein) transporter substrate inhibitor. Interactions are anticipated with anthracyclines, mitoxantrone, methotrexate, topotecan and irinotecan, imatinib and other tyrosine kinase inhibitors, irinotecan and statins (ie rosavustatin), glyburide, nitrofurantoin, dipyridamole, cimetidine, chlorothiazide, sulfasalazine, ketamine, prazosin and leflunomide which may or may not be clinically significant. Concomitant use with rosuvastatin increased rosuvastatin concentrations by about 120%. Increased exposure (often through increased absorption from the intestine) of these drugs is possible, and the dose may need adjustment. Find out more using the Antifungal Drug Interactions database and app produced by the Fungal Infection Trust. |
| SIDE EFFECTS & TOXICITY Oteseconazole in pregnant rats resulted in several different ocular abnormalities at doses about 3.5 times the steady state clinical exposure seen with patients being treated for RVVC. As a result, and especially given the drugs long half life in the body, oteseconazole is contraindicated in females of reproductive potential and in pregnancy. The most common adverse events during therapy were headache and nausea. It had slightly fewer side effects that fluconazole. |